
段阿冰团队:双金属催化和手性磷酸催化的理论研究取得新进展

成果简介
湖南大学环境科学与工程学院段阿冰课题组在双金属催化,手性磷酸催化的理论研究取得新进展,近期在Organic Chemistry Frontiers发表了3篇论文。具体如下:
全文速览
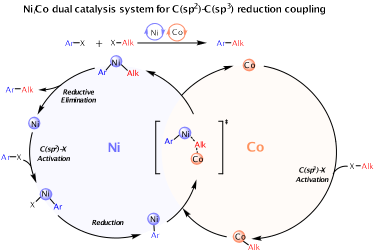
Ni/Co 双金属催化的芳基卤化物与烷基卤化物还原偶联是构建 C(sp2)−C(sp3) 键的有效策略,具有经济与兼容性优势;然而其详细反应机理长期不清晰,影响了方法学的优化与拓展。该工作构建了 Ni/Co 双金属还原偶联的完整机理框架,解释了体系的效率与兼容性来源。研究结论为优化现有 C(sp2)−C(sp3) 偶联条件与设计双金属协同催化反应提供了清晰的机理依据与方法学参考。
近日,段阿冰课题组基于 DFT 对 Ni/CoⅡ(Pc) 双催化体系进行机理解析:钴催化剂经 SN2 氧化加成活化烷基卤化物,镍催化剂则作用于芳基卤化物,随后通过反烷基化形成 C(sp2)−C(sp3) 键,并给出 Ni/Co 转金属化模型。这些见解为双催化还原偶联提供了有价值的视角并对未来双金属催化方法的发展提供了理论支撑,有望推进构建复杂分子的合成策略,并突显了双金属催化在有机合成中的潜力。第一作者为刘俊杰(2023级硕士生)、肖凤姣(2022届硕士生)、陈炜炽,通讯作者为段阿冰副教授,湖南大学环境科学与工程学院为第一单位。标题为 “Synergistic Ni/Co catalysis in C(sp2)–C(sp3) reductive coupling: a DFT study” ,DOI:10.1039/D5QO00975H
图形摘要
重要图文
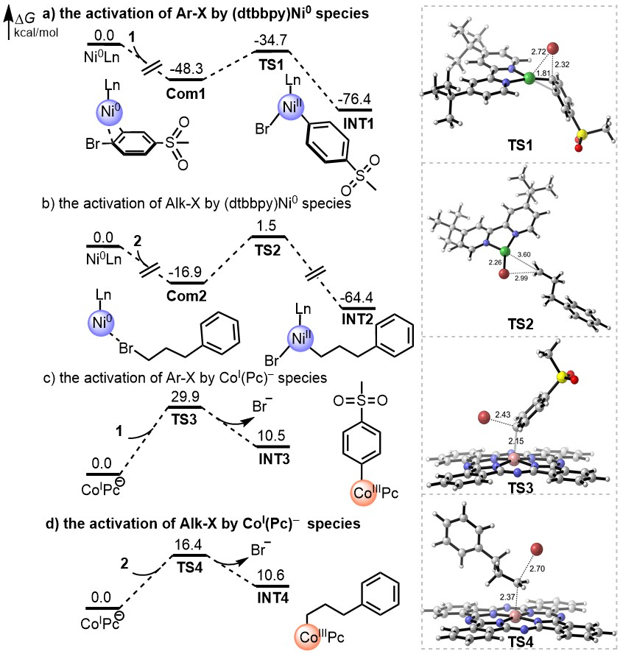
Figure 1. DFT-calculated reaction profile for the activation of 1 with 2 to Ni0Ln and CoI(Pc)-. The bond distances are given in Angstroms. Energies are in kcal/mol.
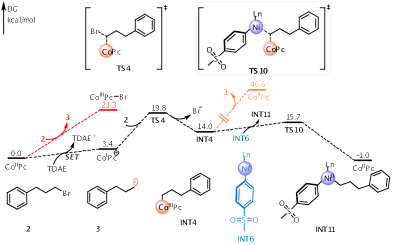
Figure 3. Calculated Gibbs energy profile for the reaction of 2 with the CoII(Pc) through SN2 oxidative addition (black) pathway and halogen-atom transfer pathway (red). Energies are in kcal/mol.

全文速览
亚甲基亚胺是一类杰出的1,3偶极化合物,是合成二氮杂环的重要组成部分,但目前手性亚甲基亚胺的合成方法依赖于动力学分解,尚不发达。本文报道了手性磷酸(CPA)催化重氮乙酸甲酯与二烯分子间[4 + 2]环加成反应的计算研究。我们的计算表明,带有给电子基团的二烯有利于[4 + 2]环加成,而CPA的窄手性口袋有利于实现优异的对映体选择性。CPA催化剂侧臂上底物与取代基之间的精细相互作用是实现优异对映选择性的必要条件。通过全面的机理研究,我们设计了一条手性偶氮亚胺的合成路线,在有机化学和药物化学领域具有很大的潜力。第一作者为陈炜炽,通讯作者为段阿冰副教授,湖南大学环境科学与工程学院为第一单位。标题为 “Chiral phosphoric acid catalyzed intermolecular [4 + 2] cycloaddition for the synthesis of chiral azomethine imines: mechanism and stereochemical model” ,DOI:10.1039/D5QO00203F
图形摘要
重要图文
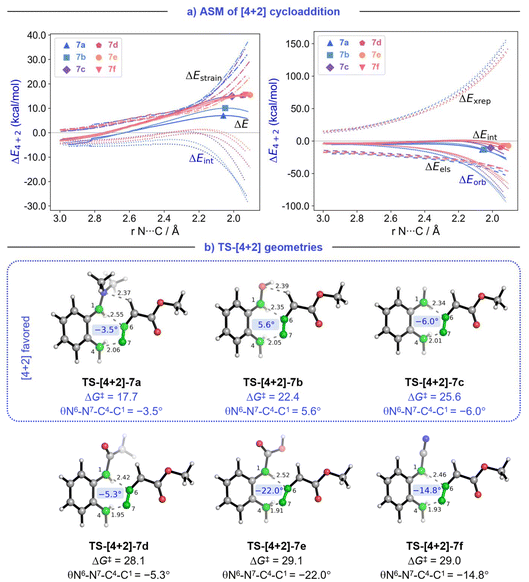
Fig. 3 (a) Activation strain analysis and energy decomposition analysis of [4 + 2] cycloaddition between MDA and dienes (7a–7f) along the reaction coordinate projected onto the N⋯C bond stretch. (b) Transition geometries and free energy of activation (ΔG‡298) (in kcal mol−1) for (4 + 2) cycloadditions between MDA and dienes (7a–7f). Bond lengths in Å.
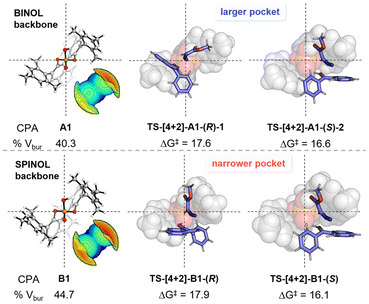
Fig. 4 Axial projections and steric maps of CPA A1 and A2, and space-filling models of transition states for [4 + 2] cycloaddition. The relative Gibbs free energy barriers for the 7bb enantiomers are given in kcal mol−1.

全文速览
非共价相互作用(NCIs)已成为不对称催化中的关键要素,在复杂转化中能够精确控制反应性和选择性。尽管它们在设计催化体系中发挥着核心作用,但对于那些同时由 NCIs 控制区域选择性和对映选择性的反应,其机理理解仍十分有限。CPA 催化的不对称反应固有的复杂性,尤其是与具有多种 NCI 活性基团的芳胺底物相关的复杂性,给理论分析和数据驱动的机理建模带来了巨大挑战。当研究对映体反转现象时,这种挑战尤为突出,因为细微的 NCI 扰动会引发不同的催化途径。至关重要的是,在多选择性反应中,独立于 CPA 基团修饰的底物控制的对映体反转在机理层面仍明显未得到充分表征。在这项研究中,我们对三种具有代表性的多位点 CPA 催化的芳胺不对称官能化反应进行了研究,通过详细的理论计算来探究空间效应和非共价相互作用的影响,以及区域选择性和对映反转之间的相互作用。本研究阐明了控制区域选择性和对映反转的因素,从而对现有框架中关键变量的机制作用进行了详细的阐释。这项工作为优化涉及芳胺的 CPA 催化的不对称反应提供了坚实的理论基础,同时也为该领域未来基于机器学习的发展提供理论依据。第一作者为李子昊,段阿冰副教授为共同通讯作者,湖南大学环境科学与工程学院为共同通讯单位。标题为 “NCI-driven regioselectivity and enantioreversal in chiral phosphoric acid-catalyzed arylamine functionalization” ,DOI:10.1039/D5QO00673B
图形摘要
重要图文
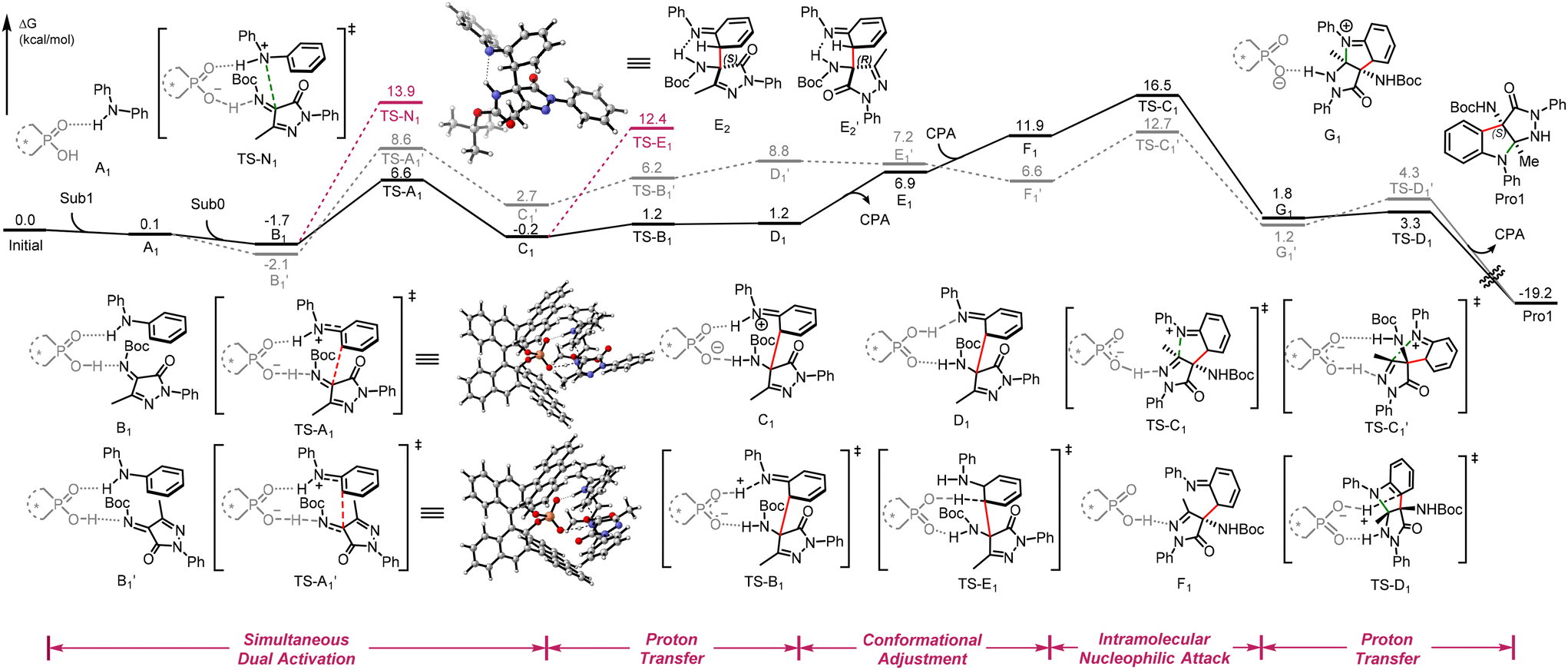
Fig. 1 Calculated Gibbs free energy profile of the reaction (1) between pyrazolinone ketimines (Sub0) and N-phenylaniline (Sub1). Energies are in kcal mol−1 and represent the relative free energies calculated in CHCl3.
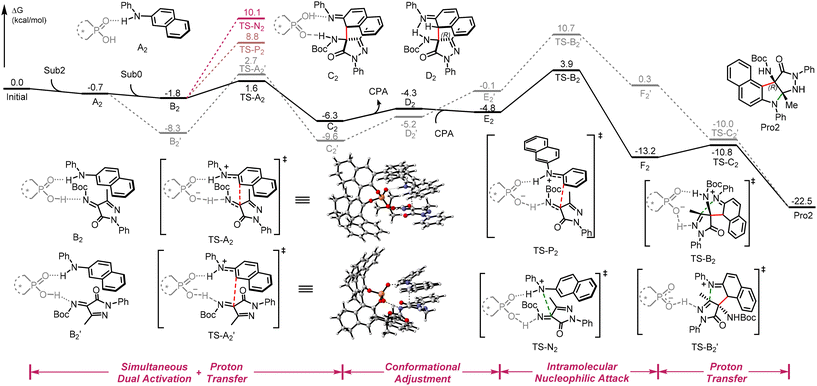
Fig. 2 Calculated Gibbs free energy profile of the reaction (2) between pyrazolinone ketimines (Sub0) and N-phenyl-2-naphthylamine (Sub2). Energies are in kcal mol−1 and represent the relative free energies calculated in DCM.
【关闭】